论文成果 | J. Phys. Chem. Lett.:中科大杨金龙、胡伟团队理论展望:二维半导体中的自旋轨道耦合效应

论文信息
英文原题:Spin-Orbit Coupling in 2D Semiconductors: A Theoretical Perspective
通讯作者:胡伟,杨金龙,中国科学技术大学
作者:陈佳佳,吴凯,胡伟,杨金龙
扫描下方二维码可阅读英文原文:

J. Phys. Chem. Lett. 2021, 12, 51, 12256–12268
Publication Date: December 20, 2021
研究背景
展望亮点
内容介绍
文中先用哈密顿模型回顾了二维半导体中Rashba和Dresselhaus效应的电子结构特征和数学表达式。
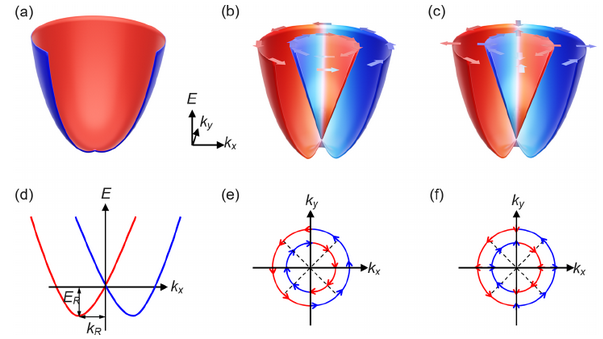
图1. Rashba效应和Dresselhaus效应的电子结构
常见的二维Rashba半导体材料可以分为以下几类。最简单的二维Rashba半导体的原胞只有2个原子,如图2a和图2c所示。第2类二维Rashba半导体是二维Janus结构,图2b和图2d展示了代表性的MoSSe和BiTeI。第3类二维Rashba半导体是二维钙钛矿(图2e)。最后,本文归纳了一些二维铁电Rashba半导体,比如图2f的扭曲1T相MoS2和图2g的AgBiP2X6。铁电极化可以实现Rashba自旋的翻转,值得进一步研究。
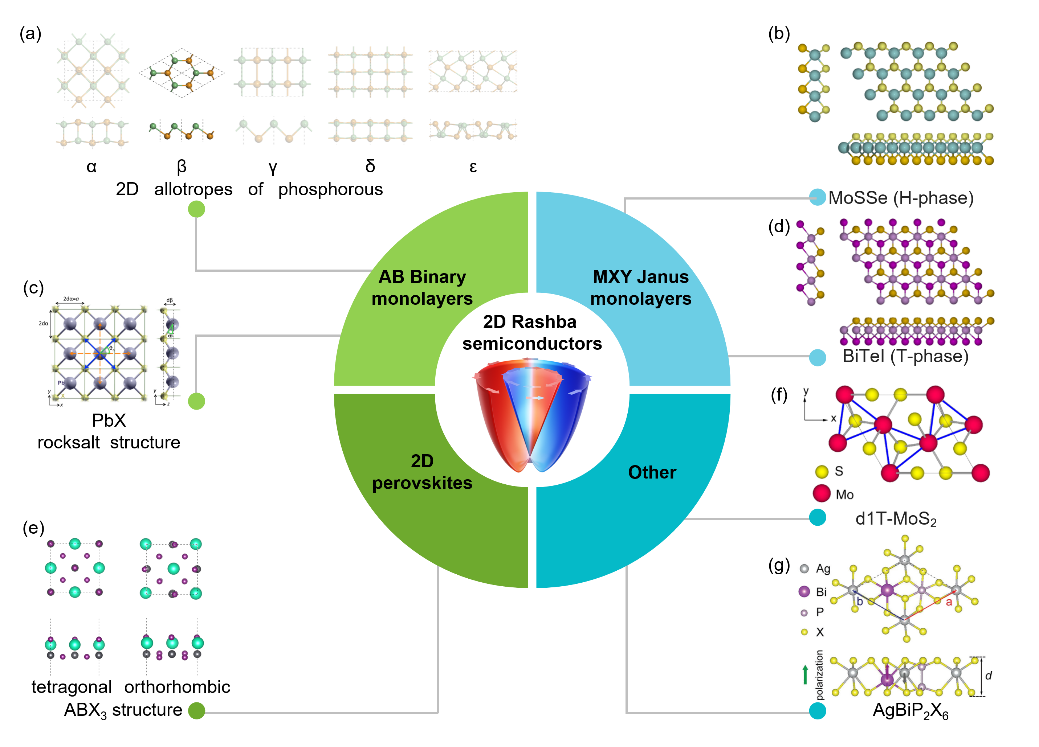
图2. 常见的二维Rashba半导体材料
考虑到实际应用,理想的Rashba材料,不仅需要有强的内禀Rashba效应,且Rashba效应该容易被调控。最常见的调控方法,是电场调控和应力调控。此外,电荷掺杂、层间相互作用(包括双层及多层结构的堆叠方式、层间距、层数)、载体的邻近效应、磁场,同样有调控作用。

图3. 调控Rashba效应的常见手段
自旋轨道耦合效应可以产生、检测和操纵自旋流,这在自旋电子学中发挥着重要作用。
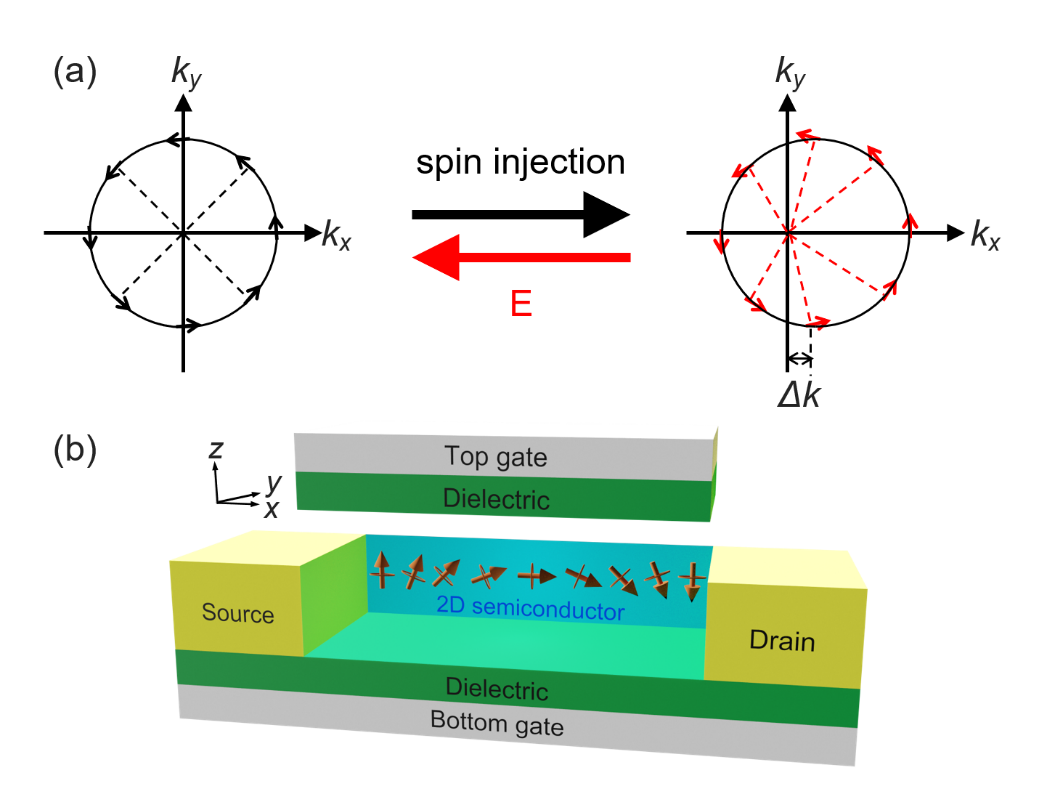
图4. Rashba效应的应用
最后,文中对自旋轨道耦合效应接下来的研究方向进行了展望:自旋轨道耦合效应的线性哈密顿量对某些材料不适用,接下来应该对非线性哈密顿量做进一步研究;二维铁电Rashba半导体,可以实现自旋的翻转,但目前的研究还比较少;一维自旋轨道耦合,包括一维Rashba效应、一维Dresselhaus效应和新型的螺旋位错自旋轨道耦合,值得深入研究,以增强对低维自旋轨道耦合效应的理解。
关于我们
上一篇:没有了下一篇:论文成果 | J. Phys. Chem. Lett.:第一性原理软件KSSOLV助力光解水催化剂理论设计


